


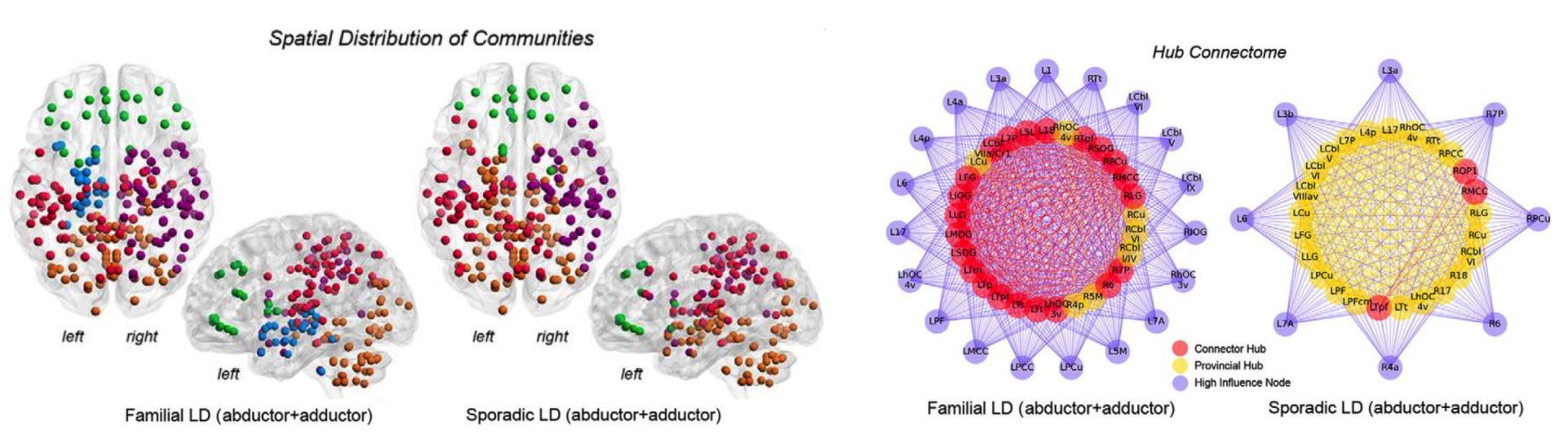





We investigate neural mechanisms of isolated focal dystonia and tremor to determine the disorder-specific pathophysiology and develop novel diagnostic tools and therapeutic strategies for these patients. We use a variety of experimental approaches, including multimodal neuroimaging, advanced machine learning, clinical-behavioral testing, neuropathology, and genetics.
Explore our website to learn more about our research, discoveries, and team.
The Dystonia and Speech Motor Control Laboratory is supported by the National Institutes of Health - National Institute on Deafness and Other Communication Disorders (NIDCD) and National Institute of Neurological Disorders and Stroke (NINDS), Department of Defense, Amazon Web Services, Jazz Pharmaceuticals, Mass General Brigham Innovation.
 Recent Publications
Recent Publications
- Failure to breathe persists without air hunger or alarm following amygdala seizures October 3, 2023
- Temporal Signature of Task-Specificity in Isolated Focal Laryngeal Dystonia July 25, 2023
- Sodium Oxybate in Alcohol-Responsive Essential Tremor of Voice: An Open-Label Phase II Study July 14, 2023
- Parkinson's disease speech production network as determined by graph-theoretical network analysis July 3, 2023
- Clinical Implications of Dystonia as a Neural Network Disorder June 20, 2023
 Dystonia and Speech Motor Control Laboratory | Department of Otolaryngology-Head & Neck Surgery, Massachusetts Eye and Ear and Harvard Medical School | 243 Charles Street, Suite 421 | Boston, MA 02114 |
Dystonia and Speech Motor Control Laboratory | Department of Otolaryngology-Head & Neck Surgery, Massachusetts Eye and Ear and Harvard Medical School | 243 Charles Street, Suite 421 | Boston, MA 02114 |